select * from intensityData limit 10;Using AWK and R to parse 25tb
big data
awk
data cleaning
Recently I was tasked with parsing 25tb of raw genotype data. This is the story of how I brought the query time and cost down from 8 minutes and $20 to a tenth of a second and less than a penny, plus the lessons learned along the way.
How to read this post: I sincerely apologize for how long and rambling the following text is. To speed up skimming of it for those who have better things to do with their time, I have started most sections with a “Lesson learned” blurb that boils down the takeaway from the following text into a sentence or two.
Just show me the solution! If you just want to see how I ended up solving the task jump to the section Getting More Creative, but I honestly think the failures are more interesting/valuable.
Intro
Recently I was put in charge of setting up a workflow for dealing with a large amount of raw DNA sequencing (well technically a SNP chip) data for my lab. The goal was to be able to quickly get data for a given genetic location (called a SNP) for use for modeling etc. Using vanilla R and AWK I was able to cleanup and organize the data in a natural way, massively speeding up the querying. It certainly wasn’t easy and it took lots of iterations. This post is meant to help others avoid some of the same mistakes and show what did eventually work.
First some background:
The Data
The data was delivered to us by our university’s genetics processing center as 25 TB of tsvs. Before handing it off to me, my advisor split and gzipped these files into five batches each composed of roughly 240 four gigabyte files. Each row contained a data for a single SNP for a single person. There were ~2.5 million SNPS and ~60 thousand people Along with the SNP value there were multiple numeric columns on things like intensity of the reading, frequency of different alleles etc. All told there were around 30 columns with frustratingly unique values.
 How a SNP Chip works. Via Research Gate
How a SNP Chip works. Via Research Gate
The Goal
As with any data management project the most important thing is to consider how the data will be used. In this case what we will mostly be doing is fitting models and workflows on a SNP by SNP basis. I.e. we only will need a single SNP’s data at a time. I needed to make it as easy, fast, and cheap as possible to extract all the records pertaining to one of the 2.5 million SNPs.
How not to do this
To appropriate a cliched quote:
I didn’t fail a thousand times, I just discovered a thousand ways not to parse lots of data into an easily query-able format.
The first attempt
Lesson Learned: There’s no cheap way to parse 25tb of data at once.
Having taken a class at Vanderbilt titled ‘Advanced methods in Big Data’ I was sure I had this in the bag. Capital B capital D Big Data, so you know it’s serious.It would be maybe an hour or two of me setting up a Hive server to run over all our data and then calling it good. Since our data is stored on AWS S3 I used a service called Athena which allows you to run Hive SQL queries on your S3 data. Not only do you get to avoid setting/ spinning up a Hive cluster, you only pay for the data searched.
After pointing Athena to my data and its format I ran a few tests with queries like
and got back results fast and well formed. I was set.
Until we tried to use the data in real life….
I was asked to grab all the data for a SNP so we could test a model on it. I ran the query:
select * from intensityData
where snp = 'rs123456';… and I waited. Eight minutes and 4+ terabytes of data queried later I had my results. Athena charges you by data searched at the reasonable rate of $5 per TB. So this single query cost $20 and eight minutes. If we ever wanted to run a model over all the data we better be ready to wait roughly 38 years and pay $50 million. Clearly this wasn’t going to work.
This should be a walk in the Parquet…
Lesson Learned: Be careful with your Parquet file sizes and organization.
My first attempt to remedy the situation was to convert all of the TSV’s to Parquet files. Parquet files are good for working with larger datasets because they store data in a ‘columnar’ fashion. Meaning each column is stored in its own section of memory/disk, unlike a text file with lines containing every column. This means to look for something you only have to read the necessary column. Also, they keep a record of the range of values by column for each file so if the value you’re looking for isn’t in the column range Spark doesn’t waste it’s time scanning through the file.
I ran a simple AWS Glue job to convert our TSVs to Parquet and hooked up the new Parquet files to Athena. This took only around five hours. However, when I ran a query it took just about the same amount of time and a tiny bit less money. This is because Spark in its attempt to optimize the job just unzipped a single TSV chunk and placed it in its own Parquet chunk. Because each chunk was big enough to contain multiple people’s full records, this meant that every file had every SNP in them and thus Spark had to open all of them to extract what we wanted.
Interestingly the default (and recomended) Parquet compression type: ‘snappy’ is not splitable. So each executor was still stuck with the task of uncompressing and loading an entire 3.5gig dataset.
Sorting out the issue
Lesson Learned: Sorting is hard, especially when data is distributed.
I thought that I had the problem figured out now. All I needed to do was to sort the data on the SNP column instead of the individual. This would allow a given chunk of data to only have a few SNPs in it and Parquet’s smart only-open-if-values-in-range feature could shine. Unfortunately, sorting billions of rows of data distributed across a cluster is not a trivial task.
Me taking algorithms class in college: "Ugh, no one cares about computational complexity of all these sorting algorithms"
— Nick Strayer (@NicholasStrayer) March 11, 2019
Me trying to sort on a column in a 20TB #Spark table: "Why is this taking so long?" #DataScience struggles.
AWS doesn’t exactly want to give refunds for the cause ‘I am an absent minded graduate student.’
After attempting to run this on Amazon’s glue it ran for 2 days and then crashed.
What about partitioning?
Lesson Learned: Partitions in Spark need to be balanced.
Another idea I had was to partition the data into chromosomes. There are 23 of these (plus a few extra to account for mitochondrial DNA or unmapped regions). This would provide a way of cutting down the data into much more manageable chunks. By adding just a single line to the Spark export function in the glue script: partition_by = "chr", the data should be put into those buckets.
 DNA is made up of multiple chunks called Chromosomes. Img via kintalk.org.
DNA is made up of multiple chunks called Chromosomes. Img via kintalk.org.
Unfortunately things didn’t work out well. This is because the chromosomes are different sizes and thus have different amounts of data within them. This meant that the tasks Spark sent out to its workers were unbalanced and ran slowly due to some of the nodes finishing early and sitting idle. The jobs did finish, however. But when querying for a single SNP the unbalance caused problems again. With SNPS in larger chromosomes (aka where we will actually want to get data) the cost was only improved ~10x. A lot but not enough.
What about even finer partitioning?
Lesson Learned: Never, ever, try and make 2.5 million partitions.
I decided to get crazy with my partitioning and partitioned on each SNP. This guaranteed that each partition would be equal in size. THIS WAS A BAD IDEA. I used Glue and added the innocent line of partition_by = 'snp'. The job started and ran. A day later I checked and noticed nothing had been written to S3 yet so I killed the job. Turns out Glue was writing intermediate files to hidden S3 locations, and a lot of them, like 2 billion. This mistake ended up costing more than a thousand dollars and didn’t make my advisor happy.
Partitioning + Sorting
Lesson Learned: Sorting is still hard and so is tuning Spark.
The last attempt in the partitioning era was to partition on chromosome and then sort each partition. In theory this would have made each query quicker because the desired SNP data would only reside in the ranges of a few of the Parquet chunks within a given region. Alas, it turns out sorting even the partitioned data was a lot of work. I ended up switching to EMR for a custom cluster, using 8 powerful instances (C5.4xl) and using Sparklyr to build a more flexible workflow…
# Sparklyr snippet to partition by chr and sort w/in partition
# Join the raw data with the snp bins
raw_data
group_by(chr) %>%
arrange(Position) %>%
Spark_write_Parquet(
path = DUMP_LOC,
mode = 'overwrite',
partition_by = c('chr')
)…but no mater what the job never finished. I tried all the tuning tricks: bumped up the memory allocated to each executor of the queries, used high ram node types, broadcasting variables, but it would always get around half way done then executors would slowly start failing till everything eventually ground to a halt.
Update: so it begins. pic.twitter.com/agY4GU2ru5
— Nick Strayer (@NicholasStrayer) May 15, 2019
Getting more creative
Lesson Learned: Sometimes bespoke data needs bespoke solutions.
Every SNP has a position value. This is an integer corresponding to how many bases along its chromosome it lies. This is a nice and natural method of organizing our data. The first thought I had was building partitions by regions of each chromosome. Aka (positions 1 - 2000, 2001 - 4000, etc). The problem is SNPs are not evenly distributed along their chromosomes, so the bins would be wildly different in size.
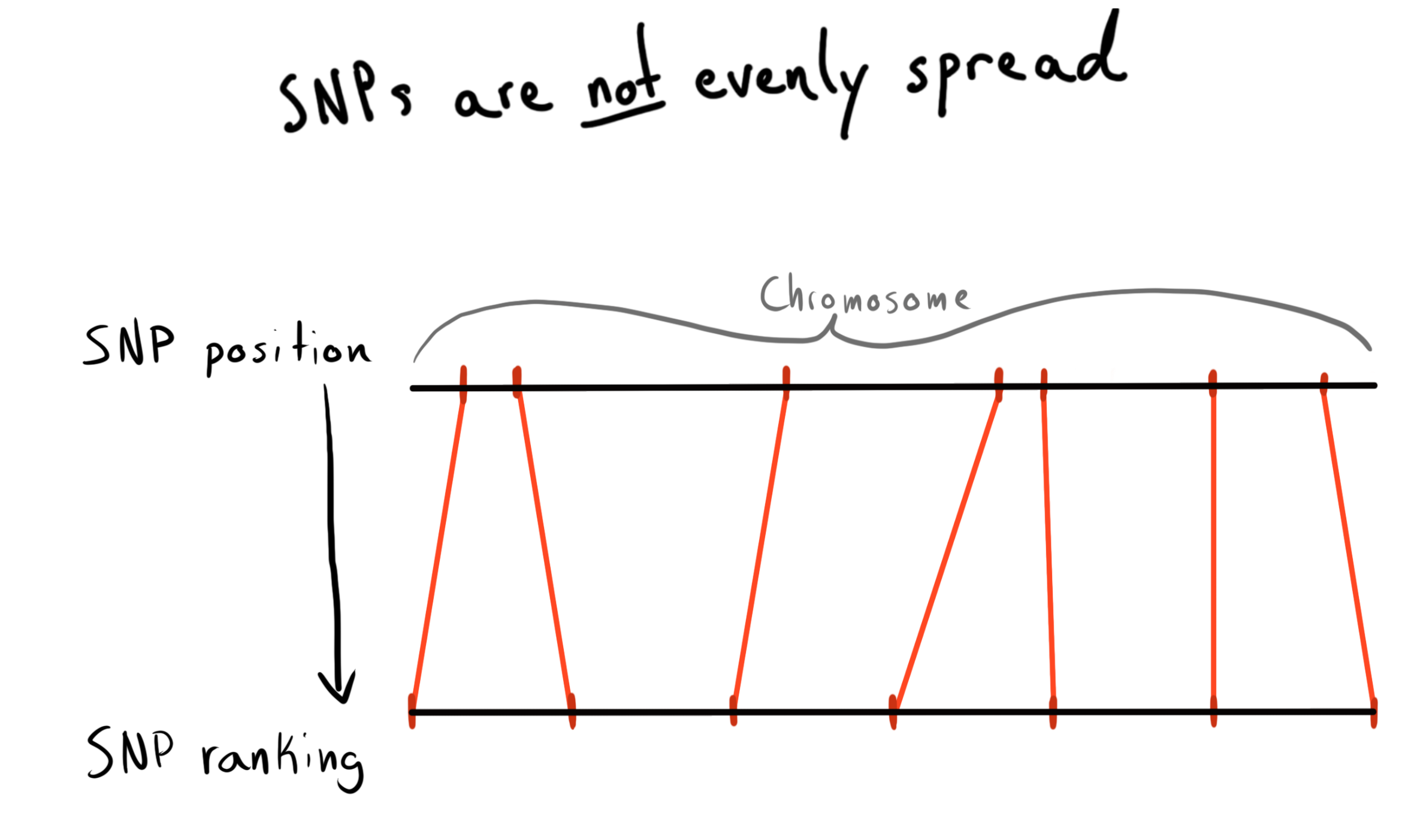
The solution I came up with was to bin by position rank. I ran a query on our already loaded data to get the list of the unique SNPs, their positions, and their chromosomes. I then sorted within each chromosome and bundled the SNPs into bins of a given size. E.g. 1000 SNPS. This gave me a mapping from SNP -> bin-in-chromosome.
I ended up using 75 SNPs per bin, I explain why later.
snp_to_bin <- unique_snps %>%
group_by(chr) %>%
arrange(position) %>%
mutate(
rank = 1:n()
bin = floor(rank/snps_per_bin)
) %>%
ungroup()First attempt with Spark
Lesson Learned: Spark joining is fast, but partitioning is still expensive
The goal was to read this small (2.5 million row) dataframe into Spark, join it with the raw data, and then partition on the newly added bin column.
# Join the raw data with the snp bins
data_w_bin <- raw_data %>%
left_join(sdf_broadcast(snp_to_bin), by ='snp_name') %>%
group_by(chr_bin) %>%
arrange(Position) %>%
Spark_write_Parquet(
path = DUMP_LOC,
mode = 'overwrite',
partition_by = c('chr_bin')
)Notice the use of sdf_broadcast(), this lets Spark know it should send this dataframe to all nodes. It’s helpful when the data is small and needed for all tasks. Otherwise Spark tries to be clever and waits to distribute it till it needs it which can cause bottlenecks.
Again, things didn’t work out. Like the sorting attempt, the jobs would run for a while, finish the joining task, and then as the partitioning started executors would start crashing.
Bringing in AWK
Lesson Learned: Don’t sleep on the basics. Someone probably solved your problem in the 80s.
Up to this point all my Spark failures were due to the data being shuffled around the cluster because it was starting all mixed up. Perhaps I could help it out with some preprocessing. I decided to try and split the raw text data on the chromosome column, that way I would be able to provide Spark with somewhat ‘pre-partitioned’ data.
I stack overflow searched how to split by column value and found this wonderful answer. Using AWK you can split a text file up by a column’s values by performing the writing in the script rather than sending results to stdout.
I wrote up a bash script to test this. I downloaded one of the gzipped tsv, then unzipped it using gzip, piped that to awk.
gzip -dc path/to/chunk/file.gz |
awk -F '\t' \
'{print $1",..."$30">"chunked/"$chr"_chr"$15".csv"}'It worked!
Saturating the cores
Lesson Learned: gnu parallel is magic and everyone should use it.
The splitting was a tad bit slow and when I ran htop to see the usage of the powerful (expensive) ec2 instance I was using a single core and ~200 MB of ram. If I wanted to get things done and not waste a lot of money I was going to need to figure out how to parallelize. Luckily I found the chapter on parallelizing workflows in Data Science at the Command Line, the utterly fantastic book by Jeroen Janssens. It introduced me to gnu parallel which is very flexible method for spinning up multiple threads in a unix pipeline.

Once I ran the splitting using the new GNU parallel workflow it was great, but I was still getting some bottle-necking caused by downloading the S3 objects to disk being a little bit slow and not fully parallelized. I did a few things to fix this.
It was pointed out on twitter by Hyperosonic that I forgot to cite gnu parallel properly as requested by the package. You would think that the number of times I saw the message reminding me to cite that wouldn’t be possible! Tange, Ole. ‘Gnu parallel-the command-line power tool.’ The USENIX Magazine 36.1 (2011): 42-47.
- Found out that you can implement the S3 download step right into the pipeline, completely skipping intermediate disk storage. This meant I could avoid writing the raw data to disk and also use smaller and thus cheaper storage on AWS.
- Increased the number of threads that the AWS CLI uses to some large number (the default is 10) with
aws configure set default.s3.max_concurrent_requests 50. - Switched to a network speed optimized ec2 instance. These are the ones with the
nin the name. I found that the loss in compute power caused from using the ‘n’ instances was more than made up for by the increased download speeds. I used c5n.4xl’s for most of my stuff. - Swapped
gziptopigz, which is a parallel gzip tool that does some clever things to parallelize an inherently unparallelizable task of decompressing gziped files. (This helped the least.)
# Let S3 use as many threads as it wants
aws configure set default.s3.max_concurrent_requests 50
for chunk_file in $(aws s3 ls $DATA_LOC | awk '{print $4}' | grep 'chr'$DESIRED_CHR'.csv') ; do
aws s3 cp s3://$batch_loc$chunk_file - |
pigz -dc |
parallel --block 100M --pipe \
"awk -F '\t' '{print \$1\",...\"$30\">\"chunked/{#}_chr\"\$15\".csv\"}'"
# Combine all the parallel process chunks to single files
ls chunked/ |
cut -d '_' -f 2 |
sort -u |
parallel 'cat chunked/*_{} | sort -k5 -n -S 80% -t, | aws s3 cp - '$s3_dest'/batch_'$batch_num'_{}'
# Clean up intermediate data
rm chunked/*
doneThese steps combined to make things very fast. By virtue of increasing the speed of download and avoiding writing to disk I was now able to process a whole 5 terabyte batch in just a few hours.
There's nothing sweeter than seeing all the cores you're paying for on AWS being used. Thanks to gnu-parallel I can unzip and split a 19gig csv just as fast as I can download it. I couldn't even get Spark to run this. #DataScience #Linux pic.twitter.com/Nqyba2zqEk
— Nick Strayer (@NicholasStrayer) May 17, 2019
This tweet should have said ‘tsv’. Alas.
Using newly parsed data
Lesson Learned: Spark likes uncompressed data and does not like combining partitions.
Now that I had the data sitting in an unzipped (see splittable) and semi-organized format on S3 I could go back to Spark. Surprise – things didn’t workout again! It was very hard to accurately tell Spark how the data was partitioned and even when I did it seemed to like to split things into way too many partitions (like 95k), which then when I used coalesce to reduce down to a reasonable number of partitions, ended up ruining the partitioning I had used. I am sure there is a way to fix this but I couldn’t find it over a couple days of looking. I did end up getting things to finish on Spark, it took a while however and my split Parquet files were not super tiny (~200KB) But the data was where it needed to be.
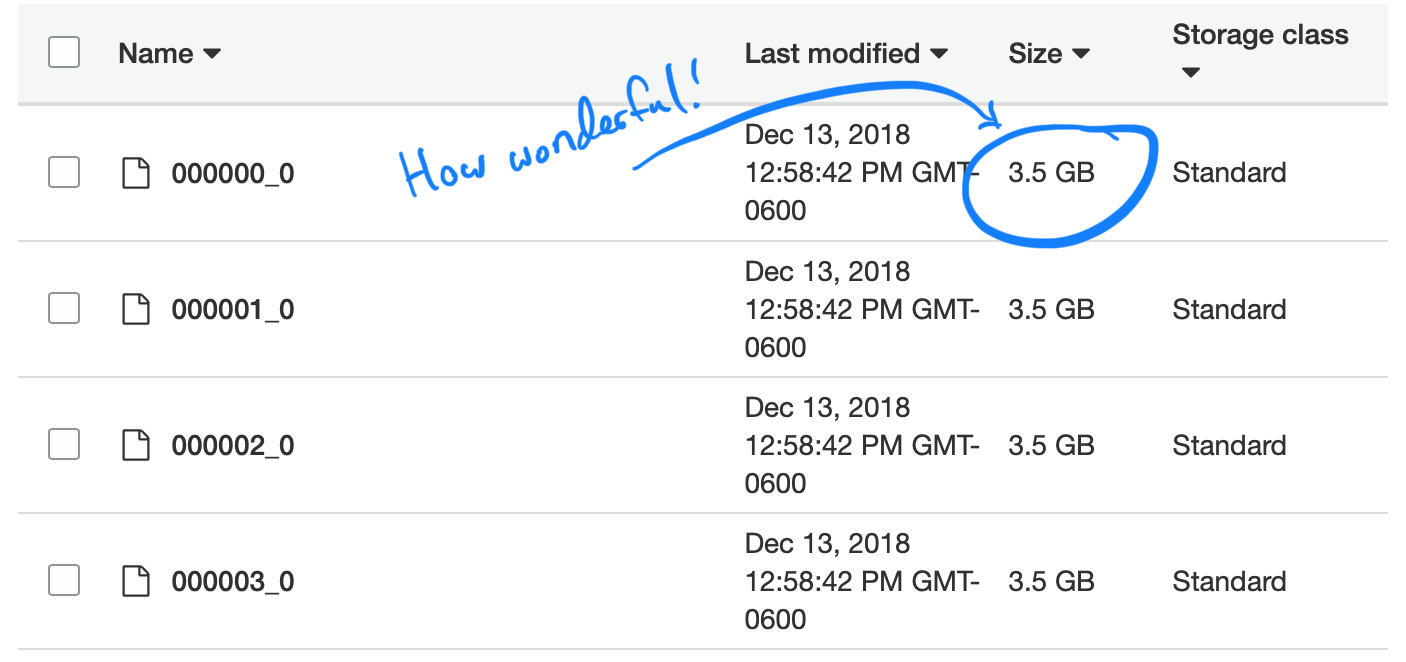
 Too small and uneven, wonderful!
Too small and uneven, wonderful!
Testing out local Spark queries
Lesson Learned: Spark is a lot of overhead for simple jobs.
With the data loaded up into a reasonable format I could test out the speed. I setup an R script to spin up a local Spark server and then load a Spark dataframe from a given Parquet bins location. I tried loading all the data but couldn’t get Sparklyr to recognize the partitioning for some reason.
sc <- Spark_connect(master = "local")
desired_snp <- 'rs34771739'
# Start a timer
start_time <- Sys.time()
# Load the desired bin into Spark
intensity_data <- sc %>%
Spark_read_Parquet(
name = 'intensity_data',
path = get_snp_location(desired_snp),
memory = FALSE )
# Subset bin to snp and then collect to local
test_subset <- intensity_data %>%
filter(SNP_Name == desired_snp) %>%
collect()
print(Sys.time() - start_time)This took 29.415 seconds. Much better than before, but still not a great sign for mass testing of anything. In addition, I couldn’t try and speed it up by enabling caching because when I tried to cache the bin’s Spark dataframe in memory Spark always crashed, even when I gave it 50+ gigs of memory for a dataset that was at this point smaller than 15.
Back to AWK
Lesson Learned: Associative arrays in AWK are super powerful.
I knew I could do better. I remembered that I had read in this charming AWK guide by Bruce Barnett about a cool feature in AWK called “associative arrays’. These are essentially a key-value stores in AWK that for some reason had been given a different name and thus I never thought too much about. It was brought to my attention by Roman Cheplyaka that the term ‘Associative Array’ is much older than ‘key-value store’. In fact, key-value store doesn’t even show up on google ngrams when you look for it, but associative array does! In addition, key-value stores are more often associated with database systems and thus a hashmap is really a more appropriate comparison here. I realized that I could use these associative arrays to perform the union between my SNP -> bin table and my raw data without using Spark.
To do this I used the BEGIN block in my AWK script. This is a block of code that gets run before any lines of data are fed into the main body of the script.
join_data.awk
BEGIN {
FS=",";
batch_num=substr(chunk,7,1);
chunk_id=substr(chunk,15,2);
while(getline < "snp_to_bin.csv") {bin[$1] = $2}
}
{
print $0 > "chunked/chr_"chr"_bin_"bin[$1]"_"batch_num"_"chunk_id".csv"
}The while(getline...) command loaded all the rows in from my bin csv and set the first column (the SNP name) as the key to the bin associative array and the second value (the bin) to the value. Then, in the { block } that gets run on every line of the main file, each line is sent to an output file that was had a unique name based upon its bin: ..._bin_"bin[$1]"_....
The variables of batch_num and chunk_id corresponded to data given by the pipeline that allowed me to avoid race conditions in my writing by making sure that every thread run by parallel wrote to its own unique file.
Because I had all the raw data split into chromosome folders from my previous AWK experiment I could now write another bash script to work through a chromosome at a time and send back the further partitioned data to S3.
DESIRED_CHR='13'
# Download chromosome data from s3 and split into bins
aws s3 ls $DATA_LOC |
awk '{print $4}' |
grep 'chr'$DESIRED_CHR'.csv' |
parallel "echo 'reading {}'; aws s3 cp "$DATA_LOC"{} - | awk -v chr=\""$DESIRED_CHR"\" -v chunk=\"{}\" -f split_on_chr_bin.awk"
# Combine all the parallel process chunks to single files and upload to rds using R
ls chunked/ |
cut -d '_' -f 4 |
sort -u |
parallel "echo 'zipping bin {}'; cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R '$S3_DEST'/chr_'$DESIRED_CHR'_bin_{}.rds"
rm chunked/*This script has two parallel sections:
The first one reads in every file containing data for the desired chromosome and divvies them up to multiple threads that spit their files into its representative bins. In order to prevent race conditions from the multiple threads writing to the same bin file, AWK is passed the name of the file which it uses to write to unique locations, e.g. chr_10_bin_52_batch_2_aa.csv This results in a ton of tiny files located on the disk (I used 1TB EBS volumes for this).
The second parallel pipeline goes through and merges every bin’s separate files into single csv’s with cat, and sends them for export…
Piping to R?
Lesson Learned: You can access stdin and stdout from inside an R script and thus use it in a pipeline.
You may have noticed this part of the bash script above: ...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R.... This line pipes all the concatenated files for a bin into the following R script… The {} in there is a special parallel technique that pastes whatever data it is sending to the given thread right into the command it’s sending. Other option are {#} which gives the unique thread ID and {%} which is the job slot number (repeates but never at the same time). For all of the option checkout the docs.
#!/usr/bin/env Rscript
library(readr)
library(aws.s3)
# Read first command line argument
data_destination <- commandArgs(trailingOnly = TRUE)[1]
data_cols <- list(SNP_Name = 'c', ...)
s3saveRDS(
read_csv(
file("stdin"),
col_names = names(data_cols),
col_types = data_cols
),
object = data_destination
)By passing readr::read_csv the variable file("stdin") it loads the data piped to the R script into a dataframe, which then gets written as an .rds file directly to s3 using aws.s3.
Rds is kind-of like a junior version of Parquet without the niceties of columnar storage.
After this bash script had finished I have a bunch of .rds files sitting in S3 benefiting with the benefits of efficient compression and built-in types.
Even with notoriously slow R in the workflow, this was super fast. Unsurprisingly the parts of R for reading and writing data are rather optimized. After testing on a single average size chromosome the job finished in about two hours using a C5n.4xl instance.
Limits of S3
Lesson Learned: S3 can handle a lot of files due to smart path implementation.
I was worried about how S3 would handle having a ton of files dumped onto it. I could make the file names make sense, but how would S3 handle searching for one?
 Folders in S3 are just a cosmetic thing and S3 doesn’t actually care about the
Folders in S3 are just a cosmetic thing and S3 doesn’t actually care about the / character.From the S3 FAQs page
It turns out S3 treats the path to a given file as a simple key in what can be thought of as a hash table, or a document-based database. Think of a “bucket” as a table and every file is an entry.
Because speed and efficiency are important to S3 making money for Amazon, it’s no surprise that this key-is-a-file-path system is super optimized. Still, I tried to strike a balance. I wanted to not need to do a ton of get requests and I wanted the queries to be fast. I found that making around 20k bin files worked best. I am sure further optimizations could speed things up (such as making a special bucket just for the data and thus reducing the size of the lookup table.) But I ran out of time and money to do more experiments.
What about cross-compatibility?
Lesson Learned: Premature optimization of your storage method is the root of all time wasted.
A very reasonable thing to ask at this point is “why would you use a proprietary file format for this?” The reason came down to speed of loading (using gzipped csvs took about 7 times longer to load) and compatibility with our workflows.Once R can easily load Parquet (or Arrow) files without the overhead of Spark I may reconsider. Everyone else in my lab exclusively uses R and if I end up needing to convert the data to another format I still have the original raw text data and can just run the pipeline again.
Divvying out the work
Lesson Learned: Don’t try to hand optimize jobs, let the computer do it.
Now that I had the workflow for a single chromosome working, I needed to process every chromosome’s data. I wanted to spin up multiple ec2 instances to convert all my data but I also didn’t want to have super unbalanced job loads (just like how Spark suffered from the unbalanced partitions). I also didn’t want to spin up a single instance for each chromosome, since there is a limit by default of 10 instances at a time for AWS accounts.
My solution was to write a brute force job optimization script using R….
First I queried S3 to figure out how large each chromosome was in terms of storage.
library(aws.s3)
library(tidyverse)
chr_sizes <- get_bucket_df(
bucket = '...', prefix = '...', max = Inf
) %>%
mutate(Size = as.numeric(Size)) %>%
filter(Size != 0) %>%
mutate(
# Extract chromosome from the file name
chr = str_extract(Key, 'chr.{1,4}\\.csv') %>%
str_remove_all('chr|\\.csv')
) %>%
group_by(chr) %>%
summarise(total_size = sum(Size)/1e+9) # Divide to get value in GB# A tibble: 27 x 2
chr total_size
<chr> <dbl>
1 0 163.
2 1 967.
3 10 541.
4 11 611.
5 12 542.
6 13 364.
7 14 375.
8 15 372.
9 16 434.
10 17 443.
# … with 17 more rowsThen I wrote a function that would take this total size info, shuffle the order, and split into num_jobs groups and report how variable the sizes of each job’s data was.
num_jobs <- 7
# How big would each job be if perfectly split?
job_size <- sum(chr_sizes$total_size)/7
shuffle_job <- function(i){
chr_sizes %>%
sample_frac() %>%
mutate(
cum_size = cumsum(total_size),
job_num = ceiling(cum_size/job_size)
) %>%
group_by(job_num) %>%
summarise(
job_chrs = paste(chr, collapse = ','),
total_job_size = sum(total_size)
) %>%
mutate(sd = sd(total_job_size)) %>%
nest(-sd)
}
shuffle_job(1)# A tibble: 1 x 2
sd data
<dbl> <list>
1 153. <tibble [7 × 3]>Once this was setup I ran a thousand shuffles using purrr and picked the best one.
1:1000 %>%
map_df(shuffle_job) %>%
filter(sd == min(sd)) %>%
pull(data) %>%
pluck(1)This gave me a set of jobs that were all very close in size. All I had do do then was wrap my previous bash script in a big for loop… It took me ~10 mins to write this job optimization which was way less time than the inballance caused by my manual job creation would have added to the processing so I think I didn’t fall for premature optimization here.
for DESIRED_CHR in "16" "9" "7" "21" "MT"
do
# Code for processing a single chromosome
fiadd a shutdown command at the end….
sudo shutdown -h now… and I was off the the races. I used the AWS CLI to spin up a bunch of instances, passing them their job’s bash script via the user_data option. They ran and then shutdown automatically so I didn’t pay for extra compute.
aws ec2 run-instances ...\
--tag-specifications "ResourceType=instance,Tags=[{Key=Name,Value=<<job_name>>}]" \
--user-data file://<<job_script_loc>>Packing it up!
Lesson Learned: Keep API simple for your end users and flexible for you.
Finally, the data was where and how I needed it. The last step was to simplify the process for using the data as much as possible for my lab members. I wanted to provide a simple API for querying. If in the future I did decide to switch from using .rds to Parquet files I wanted to be able to make that my issue and not my lab mate’s. The way I decided to to this was an internal R package.
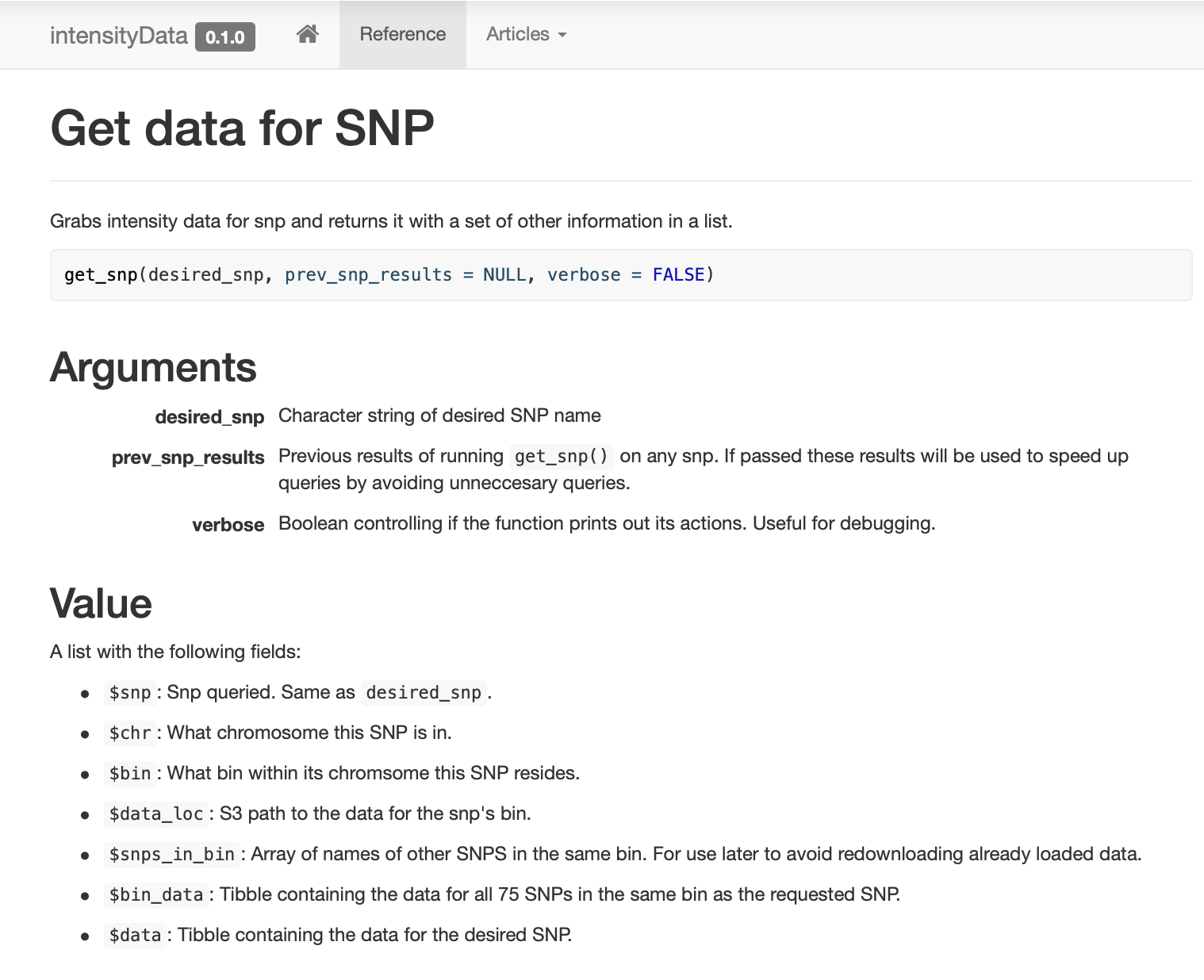
I built and documented a very simple package that contains just a few functions for accessing the data, centered around the function get_snp. Additionally, I built a pkgdown site so lab members could easily see examples/docs.
Intelligent caching.
Lesson Learned: If your data is setup well, caching will be easy!
Since one of the main workflows for these data was running the same model/ analysis across a bunch of SNPs at a time, I decided that I should use the binning to my advantage. When pulling the data down for a SNP, the entire bin’s data is kept and attached to the returned object. This means if a new query is run the old queries result’s can (potentially) be used to speed it up.
# Part of get_snp()
...
# Test if our current snp data has the desired snp.
already_have_snp <- desired_snp %in% prev_snp_results$snps_in_bin
if(!already_have_snp){
# Grab info on the bin of the desired snp
snp_results <- get_snp_bin(desired_snp)
# Download the snp's bin data
snp_results$bin_data <- aws.s3::s3readRDS(object = snp_results$data_loc)
} else {
# The previous snp data contained the right bin so just use it
snp_results <- prev_snp_results
}
...While building the package I ran a lot of benchmarks to compare the speed between different methods. I recomend it because sometimes the results went against my intuition. For instance, dplyr::filter was much faster than using indexing based filtering for grabbing rows, but getting a single column from a filtered dataframe was much faster using indexing syntax.
Notice that the prev_snp_results object contains the key snps_in_bin. This is an array of all unique SNPs in the bin, allowing fast checking for if we already have the data from a previous query. It also makes it easy for the user to loop through all the SNPs in a bin using code like:
# Get bin-mates
snps_in_bin <- my_snp_results$snps_in_bin
for(current_snp in snps_in_bin){
my_snp_results <- get_snp(current_snp, my_snp_results)
# Do something with results
}End results
We are now able to (and have started in earnest) run models and scenarios we were incapable of before. The best part is the other members of my lab don’t have to think about the complexities that went into it. They just have a function that works.
Even though the package abstracts away the details, I tried to make the format of the data simple enough that if I were to dissapear tomorrow someone could figure it out.
The speed is much improved. A typical use-case is to scan a functionally significant region of the genome (such as a gene). Before we couldn’t do this (because it cost too much) but now, because of the bin structure and caching, it takes on average less than a tenth of a second per SNP queried and the data usage is not even high enough to round up to a penny on our S3 costs.
Recently I got put in change of wrangling 25+ TB of raw genotyping data for my lab. When I started, using Spark took 8 min & cost $20 to query a SNP. After using AWK + #rstats to process, it now takes less than a 10th of a second and costs $0.00001. My personal #BigData win. pic.twitter.com/ANOXVGrmkk
— Nick Strayer (@NicholasStrayer) May 30, 2019
Take Away
This post post isn’t meant to be a how-to guide. The final solution is bespoke and almost assuredly not the optimal one. For risk of sounding unbearably cheesy this was about the journey. I want others to realize that these solutions don’t pop fully formed into peoples head’s but they are a product of trial and error.
In addition, if you are in the position of hiring someone as a data scientist please consider the fact that getting good at these tools requires experience, and experience requires money. I am lucky that I have grant funding to pay for this but many who assuredly could do a better job than me will never get the chance because they don’t have the funds to even try.
“Big Data” tools are generalists. If you have the time you will almost assuredly be able to write up a faster solution to your problem using smart data cleaning, storage, and retrieval techniques. Ultimately it comes down to a cost-benefit analysis.
All lessons learned:
In case you wanted everything in a neat list format:
- There’s no cheap way to parse 25tb of data at once.
- Be careful with your Parquet file sizes and organization.
- Partitions in Spark need to be balanced.
- Never, ever, try and make 2.5 million partitions.
- Sorting is still hard and so is tuning Spark.
- Sometimes bespoke data needs bespoke solutions.
- Spark joining is fast, but partitioning is still expensive
- Don’t sleep on the basics. Someone probably solved your problem in the 80s.
gnu parallelis magic and everyone should use it.- Spark likes uncompressed data and does not like combining partitions.
- Spark is a lot of overhead for simple jobs.
- Associative arrays in AWK are super powerful.
- You can access
stdinandstdoutfrom inside an R script and thus use it in a pipeline. - S3 can handle a lot of files due to smart path implementation.
- Premature optimization of your storage method is the root of all time wasted.
- Don’t try to hand optimize jobs, let the computer do it.
- Keep API simple for your end users and flexible for you.
- If your data is setup well, caching will be easy!